
by Jose Armando Keppis (Hostos CC, Earth System Science & Environmental Engineering, 2022-2024 CRSP cohort)
The work was done as a part of the CRSP program at Hostos Community College/CUNY, under the supervision of Dr. Yoel Rodríguez.
This article has been published as part of the Special Edition of Ad Astra, which features the CUNY Research Scholars Program (CRSP) across The City University of New York. The issue is accessible at http://www.adastraletter.com/2024/crsp-special-edition/.
ABOUT THE AUTHOR

Jose Keppis
Mr. Jose Armando Keppis is currently an Undergraduate in Earth System Science & Environmental Engineering in the City University of New York. He is currently a part of the CUNY Research ScholarsProgram (CRSP) where he is conducting research on Computational Biophysics. He has also completed research under the sponsorship of the Louis Stokes Alliances for Minority Participation (LSMAP) and Global Scholars Achieve Career Success (GSACS). He has been recognized locally and nationally for his research on COVID-19. He expects to continue his education to eventually manage and study the pollution of air, water, and waste. He dedicates his time to academic and extracurricular activities. He is currently the Co-Director of the Society of Hispanics Professional Engineers (SHPE) 29th annual Pre-College Engineering Day (PCED), a part of the Hostos Engineering Academic Talent (HEAT) Scholarship Program, Next-Gen Scholars Program, and the Black Male Initiative – Together wE Achieve More (BMI TEAM).
The global pandemic of Coronavirus Disease 2019 (COVID-19) was caused by the Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2). Viral infection occurs when the SARS-CoV-2 Spike Glycoprotein Receptor Binding Domain (RBD) attaches and fuses to the human Angiotensin-Converting Enzyme 2 (hACE2). Therapeutic drug research for SARS-CoV-2 inhibitors aims to neutralize Spike Glycoprotein to prevent infection. We hypothesize that small molecules could block the binding of SARS-CoV-2 Spike Glycoprotein RBD and hACE2. Three small-molecule inhibitors (hits) against SARS-CoV-2 Spike Glycoprotein have been identified through our group's previous research. To further this research, we conducted a ligand-guided search using vROCS 3.4.3.0 to rank around 4.3 million small molecules (eMolecules database) using the Tanimoto shape and color scoring function against hit pharmacophores. Subsequently, an analog database of 10,000 top-ranked small molecules from each hit was processed through structure-based molecular docking against the SARS-CoV-2 South Africa variant (Beta: K417N, E484K, N501Y | B.1.351) and Omicron variant (XBB.1.5) using OEDocking 4.1.1.0 and FRED 4.1.1.0 programs. The Chemgauss4 scoring function, FastROCS Toolkit, and visual inspection on VIDA 5.0.1 were utilized to determine the high-ranking molecules (~50) which are the candidates for experimentation using the SARS-CoV-2 cell-based pseudotype assay. Our goal is to sift through these small-molecule candidates to identify a therapeutic treatment for COVID-19. These post-processed small molecules have the potential to become probes that enhance the discovery of antivirals against SARS-CoV-2 and study the biology and interactions of SARS-CoV-2.
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the virus causing Coronavirus disease 2019 (COVID-19), initially caused an outbreak in Wuhan, in the province of Hubei, China, and later spread world-wide becoming a pandemic on March 11, 20201. The outbreak of the Coronavirus disease was due to the airborne transmission of SARS-CoV-22. The SARS-CoV-2 is a single-stranded RNA virus and a member of the Coronaviridae Viruses family that is also known from previous epidemics such as MERS (Middle Eastern Respiratory Syndrome) and SARS (Severe Acute Respiratory Syndrome). SARS-CoV-2 infection mechanisms are known to be mediated through these respective proteins: spike glycoprotein3, papain-like protease (PLpro)4, main proteases (Mpro)5.
Infection by SARS-CoV-2 begins once the SARS-CoV-2 Spike Glycoprotein S1 subunit, which contains the receptor binding domain (RBD), binds to the human Angiotensin Converting Enzyme 2 (hACE2)3. Once bound, the SARS-CoV-2 Spike Glycoprotein S2 subunit undergoes conformational changes within the trimer structure activated by the enzyme transmembrane protease, serine 2 (TMPRSS2), to fuse with the membrane6,7.
Previous therapeutics for tackling Coronaviridae Viruses aim to inhibit the spike glycoprotein8,9. Therapeutics like SARS-CoV-2 small-molecule spike glycoprotein Inhibitors (hits) target the spike glycoprotein S1 Subunit RBD to prevent binding to the hACE2. By neutralizing the virus from infecting cells,10 Rodríguez et al. 2022 discovered HCC 11 | HCC 4 | and HCC 11 (Figure 1A), which have presented antiviral selectivity towards SARS-CoV-2 Spike Glycoprotein.
HCC 1, HCC 4, and HCC 11 have therapeutic index values of 3.1, 4.0, and >8.2 respectively11. In assays where the hits are combined such as the case for HCC 1 and HCC 4, the therapeutic index results in a favorable value of 13.711. The aim of this research is to improve upon previous research to discover SARS-CoV-2 Spike Glycoprotein small-molecule inhibitor analogs of HCC 1, HCC 4, and HCC 11 using structure-based drug discovery computational biophysical methods. These analogs can potentially have improved biological activities, EC50 and therapeutic index values. We hypothesize that small molecules could disrupt the interaction between the SARS-CoV-2 Spike Glycoproteins RBD and the hACE2 to inhibit viral entry.
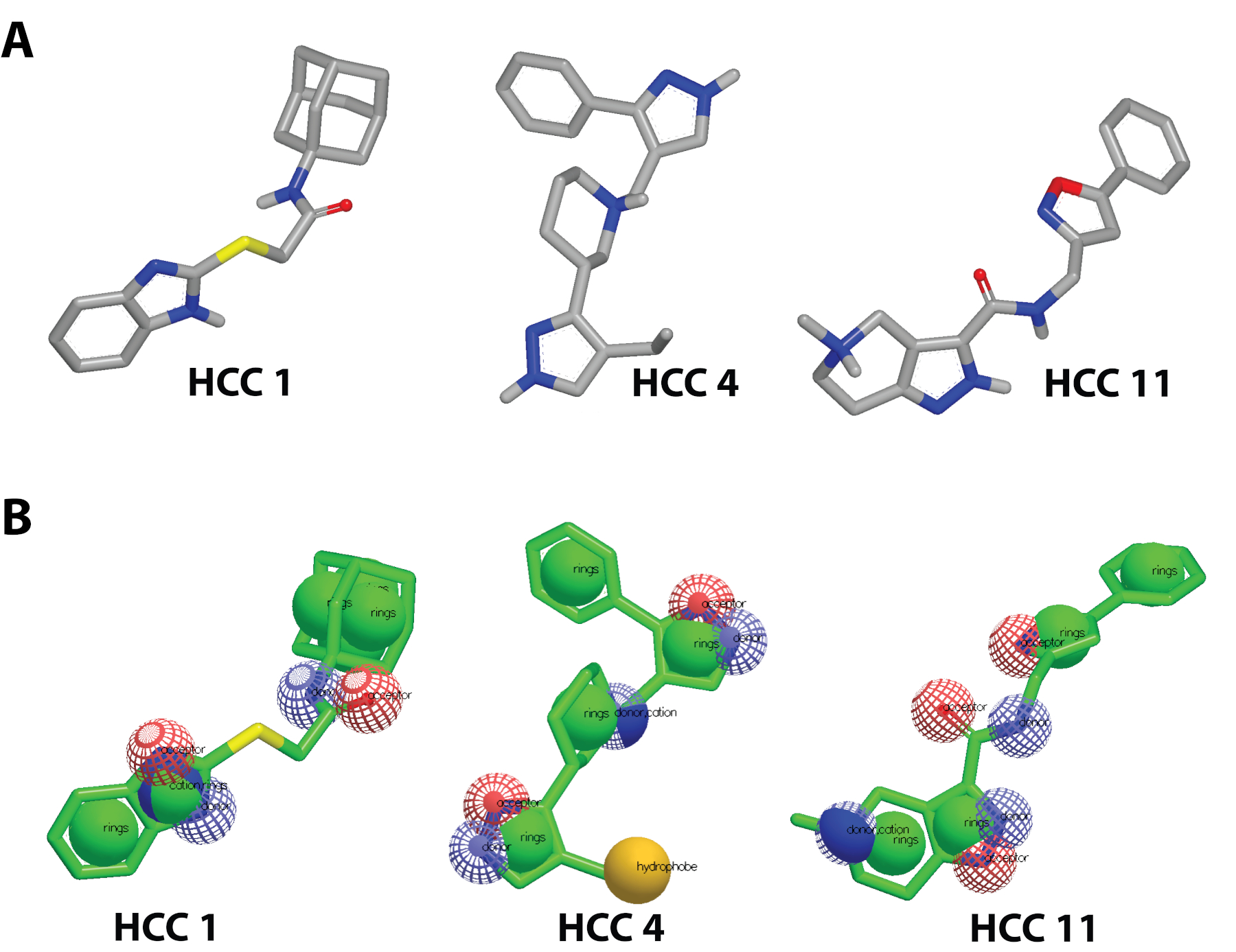
Figure 1: The 3D structures of small molecule inhibitors transformed into Pharmacophores A) Discovery of HCC 1 | HCC 4 | HCC 11 hits from Rodríguez et al. (2022)11; B) Pharmacophores of HCC 1 | HCC 4 | HCC 11 made on vROCS 3.4.3.012. All components of the structure including aromatic rings, acceptors, donors, cations, and hydrophones are labeled to model points of molecular interaction.
To conduct this research, we used a structure-based drug discovery approach to identify hit compounds through a 2D ligand-based search and a 3D ligand-based search.
The 2D ligand-based search began using the ZINC database which hosts the JSME molecule editor which is a query input tool on the Substances tab of the website13. The hit compounds’ skeletal structures were drawn onto the JSME molecule editor to search the ZINC databases on the basis of Tanimoto-40 similarity search, a mathematical model for chemical comparison in cheminfographics software’s related in shape and steric interactions. The Tanimoto coefficient measures similarity among structures represented by means of fingerprints14. Two structures with Tanimoto coefficient higher than 0.85 are considered similar. Similarly, eMolecules database molecule editor query input tool was used to conduct a similarity search based on Tanimoto-4015. Both databases resulted in a 2D analog database of hits HCC 1 | HCC 4 | HCC 11 available for purchase and virtual screening.
The 3D ligand-based search was conducted using the software suites of OpenEye Scientific16. Beginning with vROCS 3.4.3.012, a pharmacophore was made using the 3D structural information of the hits (HCC 1 | HCC 4 | HCC 11)11. The editor enabled the refinement of the interactive sites by toggling acceptors and donors. The pharmacophore is prompted as a query to the virtual screening eMolecules database (~4.3 million compounds) and the specifications of the screening (Figure 1B). 10,000 of the best potential hits were chosen to be the size of the analog database for further molecular docking16. The analog database was ranked by the TanimotoCombo similarity search that combined the ShapeTanimoto and ColorTanimoto search12.
The SARS-CoV-2 Spike Glycoprotein in complex with hACE2 (PDB ID: 7DF4) was analyzed to determine the receptor binding domain and create the docking grid box using OpenEye Make Receptor 4.1.1.017. The Box Volume was 74652 Å3 and the Dimensions were 46.00Å×44.67Å×36.33Å. An active site shape was automatically generated with a balanced contour such that the shape would not favor extending towards the solvent or the protein (Figure 2).
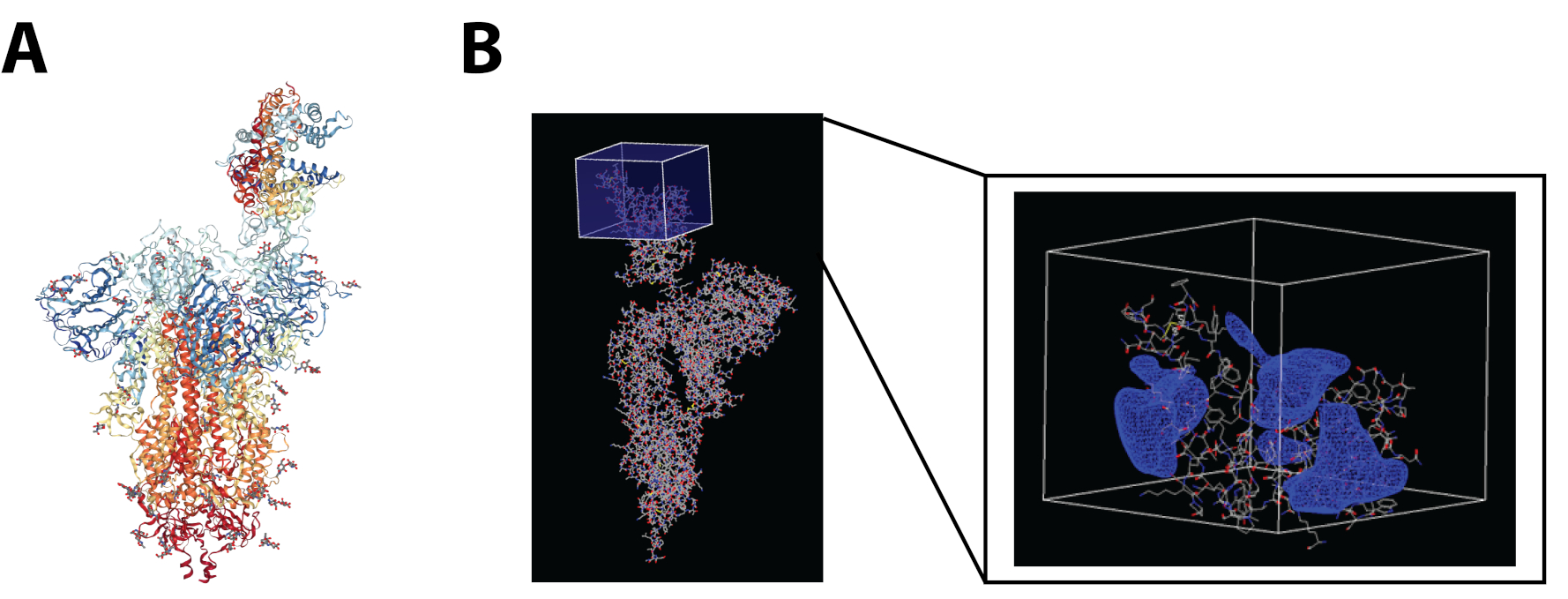
Figure 2: A) SARS-CoV-2 S Glycoprotein/hACE2 complex (PDB ID 7DF4); B) Specification of the SARS-CoV-2 S Glycoprotein RBD shown in the box and contour for the binding site generated by Make Receptor 4.1.1.017.
The contour of the SARS-CoV-2 Spike Glycoprotein RBD is flat for a binding pocket. The molecule should have a conforming shape and size to burrow or contact the contour enough to have a favorable fit. Figure 2B shows the contour as a blue mesh surrounding the RBD. Hydrogen bonding is labeled in VIDA software18 and residue interactions (ARG 403, LEU 455, TYR 453, GLN 493, TYR 495) from the three main binding pockets11 to the compounds were screened to present a diverse set of preferred molecules.
OEDocking 4.1.1.0 and Fred 4.1.1.0 program19 were then used to conduct the molecular docking of the analog molecule database to the active site of the Spike Glycoprotein receptor binding domain (PDB ID 7DF4). Fred 4.1.1.019 generated a Docking report (Figure 3) that showcased the Chemgauss4 scoring function components and main interactions. Beyond the notable scores given on the Docking report, the molecules must show traits of affinity and selectivity to the binding site. The best poses were determined by the namesake search algorithm and the generated value was described through the chemgauss4 scoring function17.
The results were generated through a docking report that presents the values as a total binding docking score, accounting for the shape, hydrogen bonds, protein desolvation, and ligand desolvation17. VIDA 5.0.1.018, a molecular visualization, and modeling platform was then used to visualize the docking models. The platform presented the best poses of the analog database molecular docking simulation to the spike glycoprotein receptor binding domain as a docked molecule structure file from FRED molecular docking. Options such as the binding contour and residue fingerprint, which are presented in the docking report, provided the necessary environment for characterizing the binding pocket, steric interactions, and candidates for further experimentation with experimental assays.
The 2D ligand-based structure searches through the ZINC13 and eMolecules15 databases, queried commercially available molecules through the Tanimoto-40 similarity search. eMolecules generated the largest selection of compounds for HCC1, HCC4, and HCC11 with 320, 18,000, and 130,000 while ZINC generated 169, 10, and 6, respectively.
The top 10,000 compounds obtained from structural molecular docking calculations from each of the pharmacophores (Figure 1) were analyzed and filtered using the Chemgauss4 scoring function. Resultant data was visualized on VIDA 5.0.1.018 to conduct an inspection of molecular interactions. Standards for experimental candidates were based on the shape within the binding pocket, its protein interaction including hydrogen bonds, aromatic interactions, and groups of molecules having the same binding pocket. Figure 3 shows an example of visual inspection and docking report20. Specifically, Figure 3A shows a docking model of a small molecule bound to SARS-CoV-2 Spike Glycoprotein RBD, which was used to guide the discovery potential SARS-CoV-2 small-molecule inhibitors. Likewise, Figure 3B shows an example of the docking report that together with molecular visualization (Figure 3A) helped to select the top fifty compounds to be assessed experimentally through in-vitro cell-based pseudotype assays. No further information of ligands is presented here to keep the specific structures of the compounds confidential.
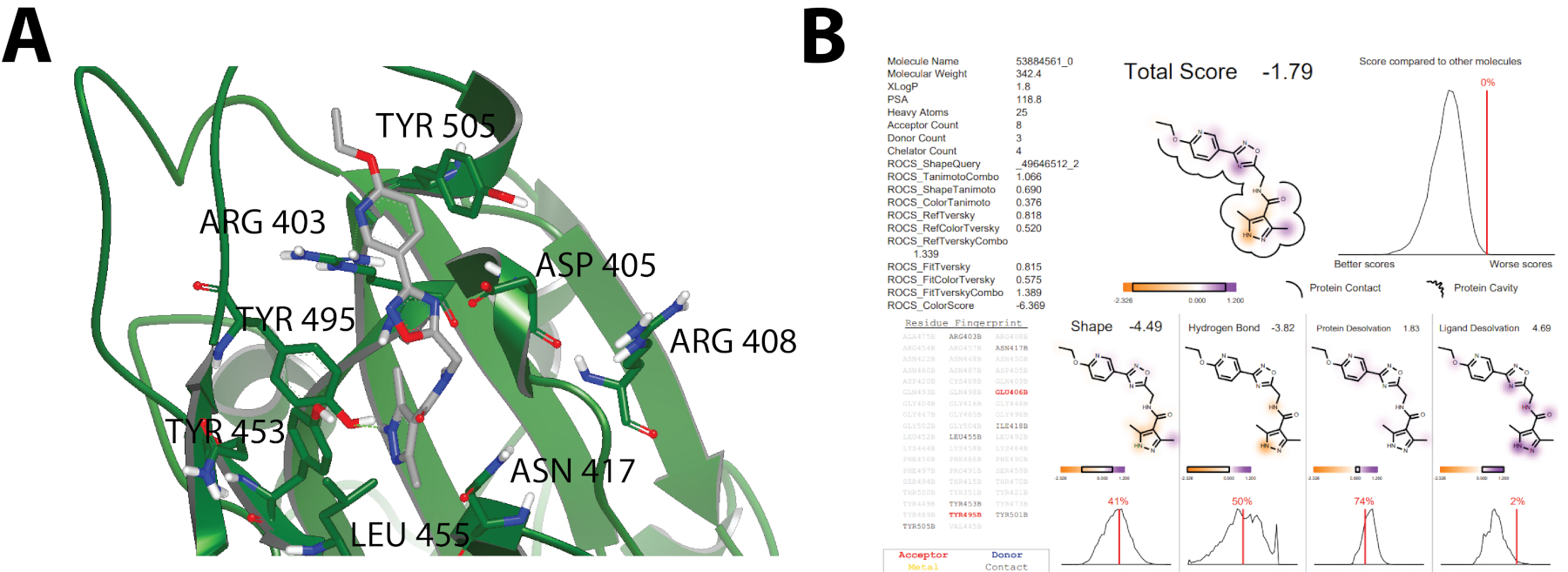
Figure 3: A) Example of visual inspection on VIDA 5.0.1.018 where a small molecule is shown in a binding pocket interacting with ARG403, ASP405, ARG408, ASN417, TYR453, LEU455, TYR495, TYR505. Binding contour is omitted. Hydrogen bonds are calculated by VIDA 5.0.1.0, steric interactions and sought out interactions including pi-pi interactions are omitted. B) The docking report summarizing the main interactions between the protein (SARS-CoV-2 Spike Glycoprotein RBD) and ligands including contact surface, residues, and binding energy via the Chemgauss4 Scoring function17.
Analogs of HCC 1 | HCC 4 | HCC 11 were investigated with the aim to discover more potent small-molecule inhibitors of SARS-CoV-2 Spike Glycoprotein with better therapeutic index. Based on the hits found by Rodríguez et al. 2022, and our current research, the combined ligand- and structure-based drug discovery is an adequate technique to predict SARSCoV-2 Spike Glycoprotein small-molecule inhibitors. Approximately fifty small-molecule inhibitor candidates against SARS CoV-2 have been identified to be purchased for in-vitro cell based pseudotype assays. These post-processed small molecules have the potential to become probes that enhance the discovery of antivirals against SARS-CoV-2 and study the biology and interactions of SARS-CoV-2.
We are grateful for the support provided by Hostos Community College and our mentor Dr. Yoel Rodríguez as well as our collaborator Dr. José Fernández Romero in the Population Council and Professor at Borough of Manhattan Community College. We also wanted to thank Edward Allen and Marian Albornoz for their support. This research is supported by CUNY Research Scholars Program (CRSP), the Louis Stokes Alliances for Minority Participation (LSAMP) Program (NSF 1826696), The Hostos Engineering Academic Talent (HEAT) Program (NSF 1833767), and CUNY Community College Research Grant (CCRG, No. 1738) Program. Special thanks to OpenEye Scientific Software for providing free academic license.
[1] Cucinotta D, Vanelli M. WHO Declares COVID-19 a Pandemic. Acta Biomed 2020, Mar 19;91(1):157-160. doi: doi: 10.23750/abm.v91i1.9397. PMID: 32191675; PMCID: PMC7569573.
[2] Zuo, Y. Y., Uspal, W. E., & Wei, T. Airborne Transmission of COVID-19: Aerosol Dispersion, Lung Deposition, and Virus-Receptor Interactions. ACS Nano 2020, 14(12), 16502–16524. https://doi.org/10.1021/acsnano.0c08484.
[3] Cong Xu et al. Conformational dynamics of SARS-CoV-2 trimeric spike glycoprotein in complex with receptor ACE2 revealed by cryo-EM. Sci. Adv. 7, eabe5575(2021).DOI:10.1126/sciadv.abe5575.
[4] Shin, D., Mukherjee, R., Grewe, D. et al. Papain-like protease regulates SARS-CoV-2 viral spread and innate immunity. Nature 2020, 587, 657–662. https://doi.org/10.1038/s41586-020-2601-5.
[5] Hu Q, Xiong Y, Zhu GH, Zhang YN, Zhang YW, Huang P, Ge GB. The SARS-CoV-2 main protease (Mpro): Structure, function, and emerging therapies for COVID-19. Med Comm 2020, 2022 Jul 14;3(3):e151. doi: 10.1002/mco2.151. PMID: 35845352; PMCID: PMC9283855.
[6] Shang J, Wan Y, Luo C, Ye G, Geng Q, Auerbach A, Li F. Cell entry mechanisms of SARS-CoV-2. Proc Natl Acad Sci U S A 2020, May 26;117(21):11727-11734. doi: 10.1073/pnas.2003138117. Epub 2020 May 6. PMID: 32376634; PMCID: PMC7260975.
[7] Hoffmann, M., Kleine-Weber, H., Schroeder, S., Krüger, N., Herrler, T., Erichsen, S., Schiergens, T. S., Herrler, G., Wu, N. H., Nitsche, A., Müller, M. A., Drosten, C., & Pöhlmann, S. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181(2), 271–280.e8. https://doi.org/10.1016/j.cell.2020.02.052.
[8] Huang Y, Yang C, Xu XF, Xu W, Liu SW. Structural and functional properties of SARS-CoV-2 spike protein: potential antivirus drug development for COVID-19. Acta Pharmacol Sin 2020, Sep;41(9):1141-1149. doi: 10.1038/s41401-020-0485-4. Epub 2020 Aug 3. PMID: 32747721; PMCID: PMC7396720.
[9] Du, L., He, Y., Zhou, Y. et al. The spike protein of SARS-CoV – a target for vaccine and therapeutic development. Nat Rev Microbiol 2009, 7, 226–236. https://doi.org/10.1038/nrmicro2090.
[10] Sethi, A., Sanam, S., Munagalasetty, S., Jayanthi, S., & Alvala, M. Understanding the role of galectin inhibitors as potential candidates for SARS-CoV-2 spike protein: In silico studies. RSC Advances 2020, 10(50), 29873–29884. https://doi.org/10.1039/D0RA04795C.
[11] Rodríguez, Y., Cardoze, S. M., Obineche, O. W., Melo, C., Persaud, A., & Fernández Romero, J. A. Small Molecules Targeting SARS-CoV-2 Spike Glycoprotein Receptor-Binding Domain. ACS Omega 2022, 7(33), 28779–28789. https://doi.org/10.1021/acsomega.2c00844.
[12] ROCS 3.4.3.0: OpenEye Scientific Software, Santa Fe, NM. http://www.eyesopen.com. Hawkins, P.C.D.; Skillman, A.G.; Nicholls, A. Comparison of Shape-Matching and Docking as Virtual Screening Tools. J Med Chem 2007, 50, 74.
[13] B. Bienfait and P. Ertl, JSME: a free molecule editor in JavaScript. J Cheminform 2013, 5:24.
[14] Maggiora G, Vogt M, Stumpfe D, Bajorath J. Molecular similarity in medicinal chemistry. J Med Chem 2014, Apr 24;57(8):3186-204. doi: 10.1021/jm401411z. Epub 2013 Nov 11. PMID: 24151987.
[15] eMolecules. eMolecules, Inc: La Jolla, CA, 2022. http://www.emolecules.com.
[16] OEDOCKING: OEDOCKING 4.3.0.3. OpenEye, Cadence Molecular Sciences, Inc., Santa Fe, NM.
[17] McGann, M. FRED and HYBRID docking performance on standardized datasets. J Comput Aided Mol Des 2012, 26, 897–906. https://doi.org/10.1007/s10822-012-9584-8.
[18] VIDA 5.0.1.0. OpenEye, Cadence Molecular Sciences, Santa Fe, NM. http://www.eyesopen.com.
[19] McGann, M. FRED Pose Prediction and Virtual Screening Accuracy. J Chem Inf Model 2011, 51(3), 578–596. https://doi.org/10.1021/ci100436p.
[20] Dock Report utility. OpenEye Scientific Software, Santa Fe, NM, USA http://www.eyesopen.com.