
by Dmitry Y. Brogun, Ani Iremashvili, and Stefan Valdez (Kingsborough, Biology, 2022-2023 CRSP cohort)
The work was done as a part of the CRSP program at Kingsborough Community College/CUNY, under the supervision of Dr. Dmitry Y. Brogun.
This article has been published as part of the Special Edition of Ad Astra, which features the CUNY Research Scholars Program (CRSP) across The City University of New York. The issue is accessible at http://www.adastraletter.com/2024/crsp-special-edition/.
ABOUT THE AUTHORS

Dmitry Y. Brogun
Dmitry Y. Brogun is a molecular evolutionary biologist with over a decade of experience in teaching and research. He holds a Master of Arts in ethology from Brooklyn College. He earned his Master of Philosophy and Doctor of Philosophy in Molecular, Cellular, and Developmental Genetics from the Graduate Center (CUNY). Dr. Brogun received his graduate research training from CUNY and Michigan State University and his postdoctoral research training from CUNY and Rockefeller University. Since 2007, he has been educating students in general biology, microbiology, anatomy, and physiology at CUNY. His research interests include molecular evolutionary biology and algology, emphasizing phylogenetics. Recently, he received an NSF allocation grant that allowed him to implement and pilot an online course where students performed pathogen surveillance in public metagenomic datasets collaboratively with individuals from CUNY, SDSU, NIH, NLM, and NCBI. At Kingsborough Community College, Dr. Brogun also serves as a Principal Investigator for the Metagenomics Discovery Program (MDC). He also mentors research students in the CUNY Scholar Research Program (CRSP), Collegiate Science Technology Program (CSTEP), and Bridges program.

Stefan Valdez
Stefan Valdez is a junior at Lehman College, completing a Bachelor of Science in Biological Anthropology. He has previously obtained an Associate of Science in Biology from Kingsborough Community College. Stefan is deeply interested in human evolution and hopes to pursue research within genetic anthropology, a field that uses molecular biology to advance our understanding of human evolution. Besides his passion for research, Stefan has worked as a Seasonal Field Science Assistant at The Hudson River Park Trust, where he assisted in implementing the Park’s estuarine research that helps protect and restore wildlife within the Hudson River. He is currently a Support Scientist at Biobus, where he teaches biology lessons to students k-12 in a mobile laboratory around New York City. At the core of Stefan’s work is a passion for science communication. He loves to share the joy of science and aims to make science more accessible to those within and outside the scientific community.

Ani Iremashvili
Ani Iremashvili is a Kingsborough Community College graduate in Biological Sciences. She has an extensive medical background working in various settings, including laboratories. During her studies in Kingsborough, she worked as an Emergency Medical Technician (EMT) and Medical Assistant (MA), where she was exposed to the medical world and patient care hands-on. In addition, Ani was interested in Mathematics and started volunteering to help high school students with exams such as SAT and ACT. However, Ani’s primary interest is microbiology, especially bacteriology and mycology. In the future, Ani plans to attend a four-year university.
During this study, we used DNA Metabarcoding to identify prokaryotic life within composted and non-composted soil samples from Kingsborough Community College Urban farm using 16s rRNA primers focusing on the V4 hypervariable region. The bioinformatics pipeline utilizes the Purple Line of the DNA Subway, a GUI-based version of the QIIME 2 pipeline used to analyze and classify sequencing results. We hypothesized that composted sediment would exhibit a higher microbial variation than non-composted sediment. Furthermore, specific microbial taxa found in composted soil may indicate beneficial effects on plant growth. The results showed predominant phyla across all samples, including Acidobacteria, Actinobacteria, Proteobacteria, Verrucomicrobia, Bacteroidetes, and Firmicutes. The most significant variation among samples of the two treatments was the bacteria concentrations of the eight most abundant taxa. The composted samples contained greater concentrations in all but one taxa: Saprospirae, an order of bacteria in the Phylum Bacteroidetes. This study provides information on the microbial composition of composted soil and reveals the differences in bacterial diversity among composted and non-composted soil. Our results could be used to develop better practices for soil enrichment.
Often used as a mitigation tool for cities' environmental issues, urban agriculture is shifting urban ecology (Yuan, G. N. et al., 2022). Microorganisms like bacteria are responsible for fundamental ecosystem processes like nutrient cycling, decomposition of organic matter, and stimulation of plants. For these reasons, studying and understanding the microbiomes in composted soil is essential. Composting is the aerobic process of breaking down organic matter, often by bacteria and earthworms, into a natural fertilizer that urban farmers can use as an alternative to harmful chemical fertilizers to enrich soil and plants. Bacteria hold a crucial role in the nitrogen conversion process in compost by converting atmospheric Nitrogen (N2) into Ammonium (NH4+) via ammonification and then into Nitrites (NO2-) and Nitrates (NO3-) via nitrification, which can be readily used by plants (Maeda, K. et al., 2011). Although denitrification is a vital part of the Nitrogen cycle as it releases Nitrogen back into the atmosphere, it is usually a detrimental process in urban agriculture because it results in the loss of fixed nitrogen, which is essential for building nucleic acids, amino acids, and chlorophyll in plants. Finally, denitrifying bacteria converts nitrates back to atmospheric Nitrogen so the cycle can continue. This process is vital for plants and animals to obtain Nitrogen, a significant element of life used to build amino acids and chlorophyll in plants.
This study aimed to determine the bacterial diversity among composted and non-composted soil samples obtained from Kingsborogh Community College’s urban farm (Figure 1A). We hypothesized that composted soil samples would contain greater bacterial diversity because compost provides a nitrogen-rich environment for more bacteria to thrive. Triplicate samples were collected from a compost plot, a non-compost plot, and a non-agriculture plot of soil as a control (Figure 1B). DNA from soil samples was isolated, and the 16s rRNA gene was amplified, sequenced, and processed through a data informatics pipeline to create a graphical representation of the data.
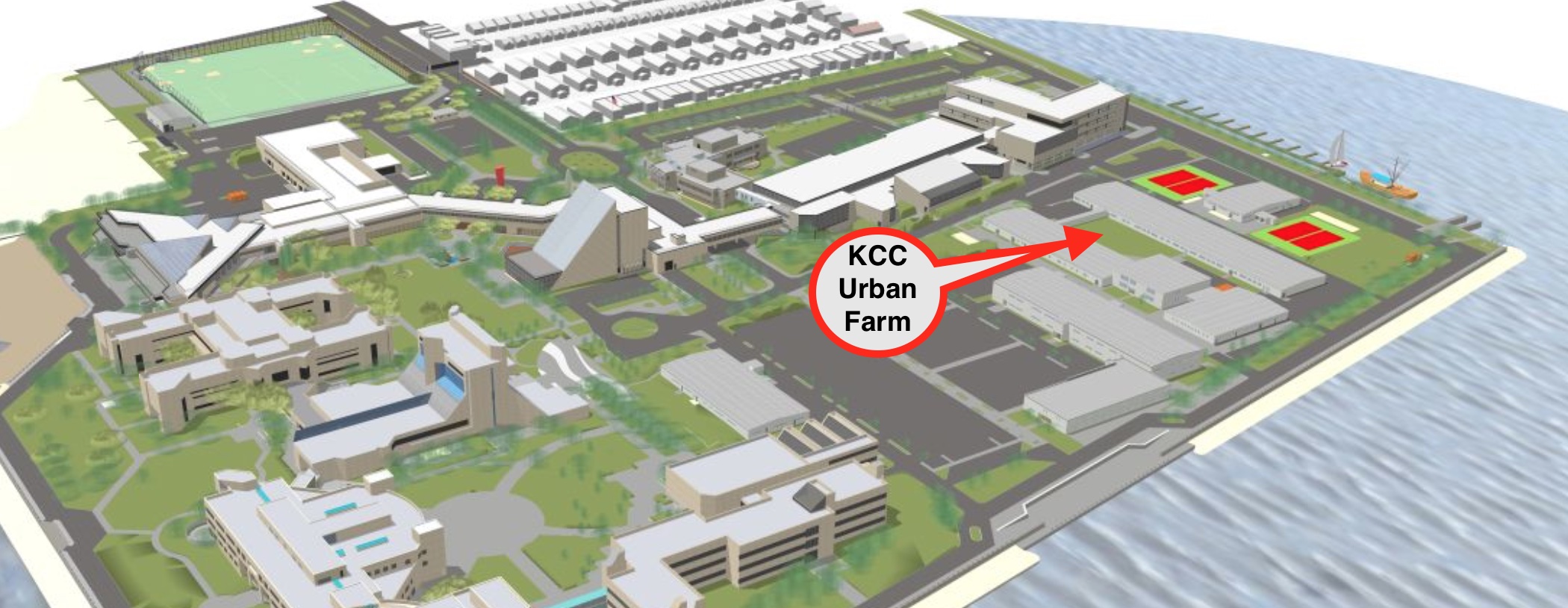
Figure 1A: A map showing Kingsborough Urban Farm's location in Brooklyn, NYC. The red arrow indicates the location of the KCC Urban Farm on the KCC campus and soil collection sites.

Figure 1B: Image of the composted, non-composted, and non-agricultural soil plots at KCC Farm. Composted and non-composted soil is depicted in the raised-bed farming plots surrounded by non-agricultural soil. Composted plots on the left two rows.
Soil samples were collected from the composted, non-composted, and non-agriculture plots into 50 ml sterile Falcon tubes from 12 centimeters beneath the organic layer in the KCC (Kingsborough Community College) urban farm, Brooklyn, NY, United States; latitude 40.5798079, longitude -73.9336462; Figure 1. Approximately 15 grams of soil were collected in triplicate at each collection site. After collection, tubes were capped with an airtight closure and transported to the Kingsborough Laboratory for DNA extraction.
The DNA from all soil samples was extracted one hour after collection. The DNA isolation protocol adapts the 2014 MO BIO DNeasy PowerSoil Pro Kit QIAGEN protocol. The protocol comprises a novel and proprietary method for isolating microbial and other DNA from environmental samples. First, 250 mg of soil were added into the powerbead pro tubes. The tubes were mixed using a vortex and 800 μl of CD1 solution, a cell membrane lysis solution. The manufacturer’s instructions were followed for the remaining DNA purification steps.
After the DNA extraction was completed, the 16S rRNA gene variable region V4 (Gray et al., 1984) was amplified using a thermocycler and barcoded oligonucleotides that contain the priming sequences 515F-GTGYCAGCMGCCGCGGTAA (Parada et al., 2016) and 806R-GGACTACNVGGGTWTCTAAT (Apprill et al., 2015). Polymerase chain reaction (PCR) was performed using the HotStarTaq Plus Master Mix Kit (Qiagen, Hilden, Germany). The thermocycling protocol was as follows: polymerase activation by heating at 94°C for 3 min; 36 cycles of denaturing at 94°C for 45 s; annealing at 50°C for 60 s; and primer extension at 72°C for 105 s. An additional elongation step of 72°C for 10 min was added to the last cycle. PCR products were run on a 2% agarose gel for the DNA amplicon visualization and purification. The library was sequenced with an Illumina MiSeq at Cold Spring Harbor Laboratory. After sequencing, barcodes were removed. The analysis of metabarcoding data sets was conducted by the Purple Line of the DNA Subway platform developed by the DNA Learning Center at Cold Spring Harbor Laboratory (https://dnasubway.cyverse.org) for the CyVerse (formerly iPlant Collaborative), an NSF-funded project headquartered at the University of Arizona. CyVerse has beta-released the Purple Line of its DNA Subway (CyVerse, 2019), a GUI-based version of the QIIME 2 pipeline (Bolyen et al., 2019).
As previously mentioned, our research aimed to determine if there were different concentrations of microbiomes in composting versus non-composting sediments due to differential urban farming techniques. Figure 2 shows the gels that reveal the amplification of the 16S rRNA gene, which was used to assess the taxonomic diversity and approximate population counts of the microbial communities in the samples. Post-sequencing, the data was analyzed through a bioinformatics pipeline utilizing the Purple Line of the DNA Subway platform (CyVerse, 2019), a GUI-based version of the QIIME 2 pipeline (Bolyen et al., 2019) and generated graphical representations of microbial data. Samples taken from various urban farm sediments contained similar phyla. The predominant phyla included Acidobacteria, Actinobacteria, Proteobacteria, Verrucomicrobia, Bacteroidetes, and Firmicutes.
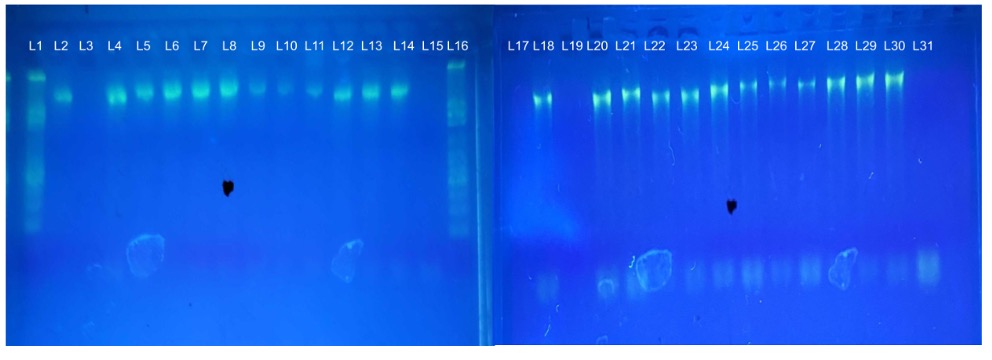
Figure 2: Gel Electrophoresis of the 16s rRNA. (L1 & L16 is the DNA Kb ladder marker (100bp-3000bp), L3, L19 & L31 correspond to the control, L2, L4 to L14 correspond to composted, and L18, L20 to L30 non-composted 16s rRNA PCR amplicon product.
Before the next-generation sequencing, a 2% gel electrophoresis was run on the PCR products. L3, L19, and L31 correspond to the negative control to ensure no contamination. All other samples showing the DNA bands for the 16s rRNA region were gel purified and sent to the Cold Spring Harbor Laboratory for next-generation sequencing.

Figure 3: Phyla concentrations in different soil samples. The taxonomic plot above represents phyla concentrations in different soil samples. Bolded boxes highlight 8 out of 758 sequenced taxa with the most significant microbial variation; see legend to the right.
The next-generation sequencing data analysis identified 758 taxa. All soil samples (composted, non-composted, and control) contained similar phyla. The predominant phyla across all samples included Acidobacteria, Actinobacteria, Proteobacteria, Verrucomicrobia, Bacteroidetes, and Firmicutes. The most significant variation was within bacteria concentrations of the first eight identified taxa, highlighted to the right in the legend.
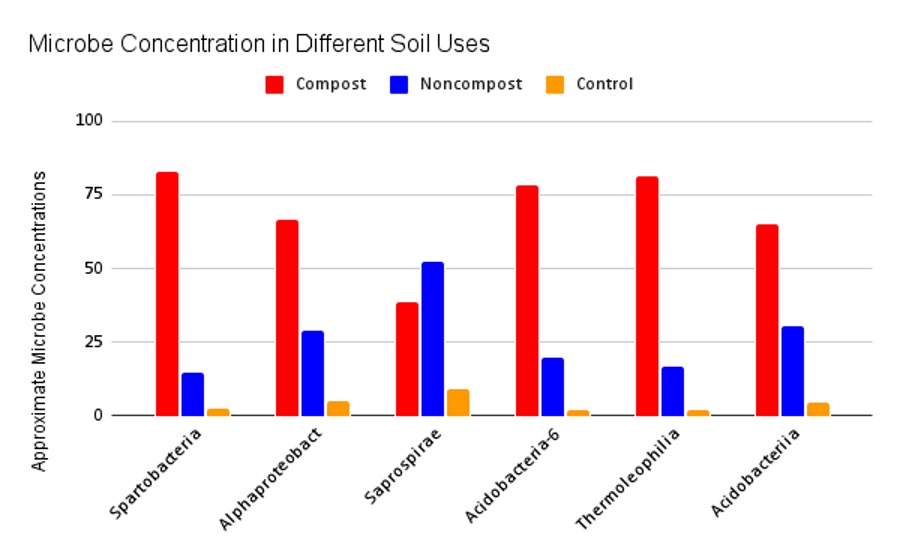
Figure 4: Microbe concentrations in different soil samples. Results from the bar graph support substantial variation in microbial concentration between the composted and non-composted samples. Most notably, the composted samples contained greater concentrations in all but one taxa, Saprospirae, an order of bacteria in Phylum Bacteroidetes. The six taxonomic orders were chosen from the legend in Figure 3.
The most significant variation among concentrations was present in the most abundant taxa (Figure 3). These taxa belong to the Phyla Verrucomicrobia, Proteobacteria, Bacteroidetes, Acidobacteria, and Actinobacteria. Six taxonomic orders were chosen and highlighted in the bar graph above to analyze their concentration in each sample. The composted samples contained greater concentrations in all but one taxa: Saprospirae, an order of bacteria in Phylum Bacteroidetes (Figure 4).
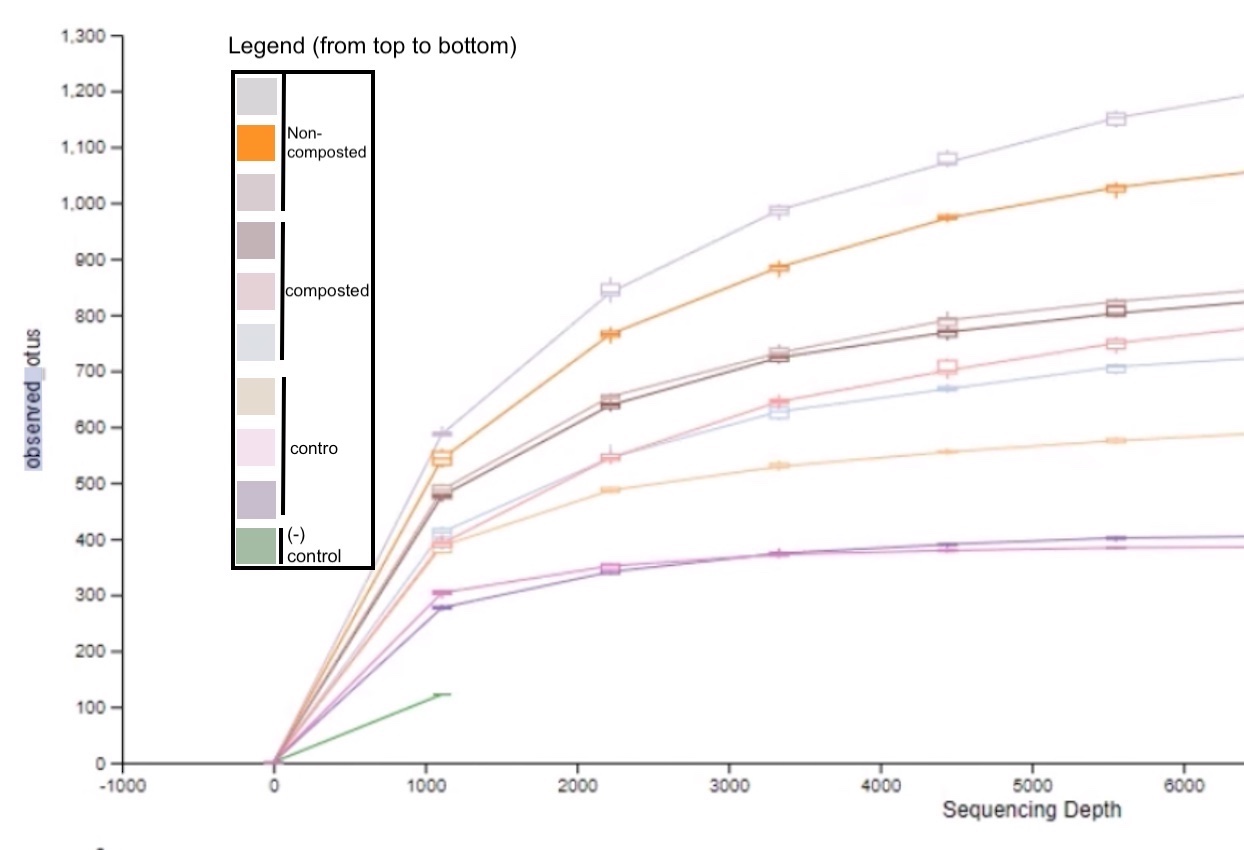
Figure 5: Microbial species richness of sediments from different urban farm beds.
The rarefaction chart data indicates that the non-composted urban farm sediments had the highest alpha diversity while the control sediments contained the lowest (Figure 5). Alpha diversity is a measure of microbial diversity within a specific location, which indicates species richness.
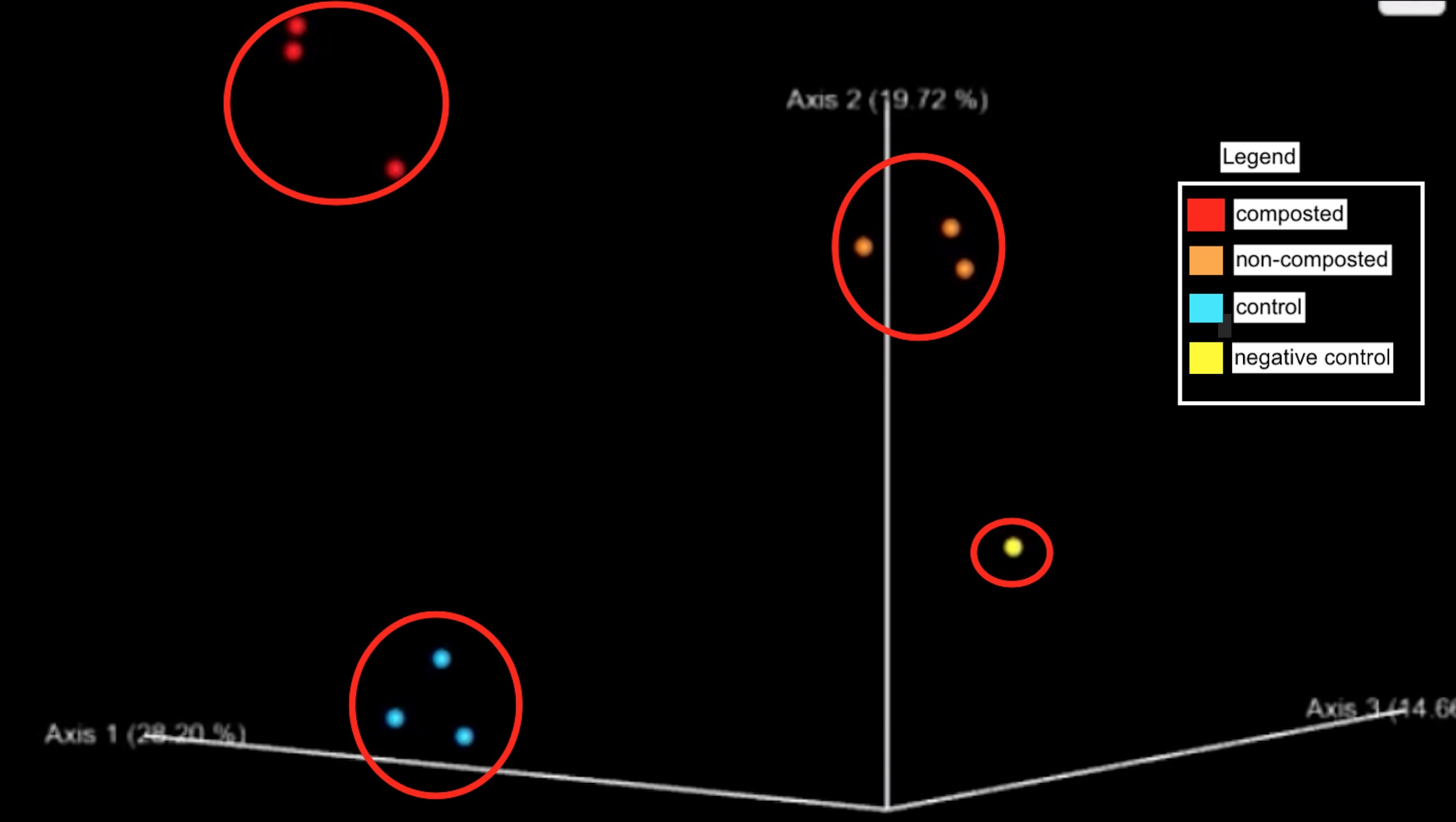
Figure 6: Principle component analysis with clusters of microbes from composted, non-composted, and controlled urban farm soil.
The considerable distance between the three sample sites on the principle component analysis (PCoA) plot indicates differences in the beta diversity between the non-composted microbial community and the composted sample. Furthermore, it is essential to note that the negative lab control is distinctly separate from all samples, supporting the validity of the results (Figure 6).
Our research aimed to analyze and compare the bacterial diversity in composted and non-composted soil from the urban farm at Kingsborough Community College. Composted soil was hypothesized to contain greater bacterial diversity than non-composted soil samples because of the nitrogen-rich environment compost provides to various bacteria. Our data indicates that the composted soil had a lower bacterial diversity than non-composted soil. Yet, we found variation in the relative abundance of phyla, not diversity (Figure 3). Next-generation sequencing of the soil samples identified 758 taxa. All soil samples (composted, non-composted, and control) contained similar phyla. The predominant phyla across all samples included Acidobacteria, Actinobacteria, Proteobacteria, Verrucomicrobia, Bacteroidetes, and Firmicutes. Non-compost and control samples had a greater diversity of taxa at the level of Order than the composted samples. However, the composted samples presented a higher abundance of the same taxa (Figure 3). Non-composted soil samples had larger species richness and significantly, species evenness than the composted soil. As per alpha diversity, the sediments of the non-composted soil appear to have the highest microbial species richness, while the control contains the lowest (Figure 5). As per beta diversity, the non-composted and composted samples are clustered a bit closer, suggesting similarities between the microbes in those two communities while maintaining distinct microbiomes. (Figure 6).
While composted samples did not have higher bacterial diversity, they did contain higher concentrations of bacteria overall (Figure 4). One outlier in this trend is Saprospirae, an order of bacteria in Phylum Bacteroidetes responsible for the degradation of complex carbohydrates like chitin. This is surprising as the degradation of complex carbohydrates is a significant process necessary in compost. One study found a negative correlation between Saprospirae and soil depth in fallow soils, indicating a decrease in Saprospirae as soil depth increased (He, et al., 2017). All of our samples were taken at 12 centimeters from the organic surface, but there was no standardized depth between all samples; therefore, this correlation should be considered.
Additionally, the composting process occurs in approximately four phases of varying temperatures: 1) the mesophilic phase (10-42°C) that lasts several days, 2) the thermophilic phase (45-70°C) that can last several months, 3) the middle mesophilic phase (65-50°C) that allows for the return of heat-resistant microbes, 4) the final phase (50-23°C) that stabilizes and matures the compost and can last several months (Papale, Maria., et al 2021). These varying stages contain different predominant communities of microorganisms. Psychrophilic bacteria are most active during the beginning and end stages when temperatures reach approximately 12°C Mesophilic bacteria thrive in moderate temperatures as the compost heats up and cools down around 20-40°C and thermophilic bacteria take over at temperatures over 40°C and can survive temperatures up to 71°C (Trautmann, N., & Olynciw, E n.d.). One of the highest concentrations of bacteria found in the compost was Thermoleophilia, an order of bacteria in the phylum Actinobacteria (Figure 4). Thermoleophilia thrives at higher temperatures, around 28-60°C Saprospirae is a mesophilic bacteria and thrives in lower temperatures of 6-47°C This indicates that the concentration of bacteria found in the composted samples is heavily influenced by the phase in which the samples were taken; it is possible that the compost was in the thermophilic phase, resulting in higher concentrations of thermophilic bacteria.
According to the data presented, bacterial communities in composted vs. non-composted soil are diverse. Composted soil was hypothesized to contain greater bacterial diversity than non-composted soil samples because of the nitrogen-rich environment compost provides to various bacteria. However, variations in the soil's microbial communities were mostly due to differences in abundance of similar bacteria, not their diversity. Overall, the composted soil contained lower bacteria diversity than the non-composted soil but a higher abundance of the same bacteria. This may be attributed to various factors, such as collection errors in soil depth or the timing of sample collection with respect to the composting phases. Additional research focusing on microbial abundance at different composting phases would unravel the factors behind the diversity of microorganisms in composted soils and their implications for urban agriculture.
The authors would like to thank Dr. Joanne Russell, Dr. Farshad Tamari, Frances Samuel, CRSP CUNY, Dr. Ron Nerio, Veer Shetty, Joshua Barnes, KCC Urban Farm Dr. Shannon Caravello and Alona Lensky, Dr. Bruce Nash, Cold Spring Harbor Laboratory DNA Learning Center.
Apprill, A., McNally, S., Parsons, R., Weber, L. (2015). Minor revision to V4 region SSU rRNA 806R gene primer greatly increases detection of SAR11 bacterioplankton. Aquat. Microb. Ecol., 75, 129–137. Available online: DOI:10.3354/ame01753.
Bolyen, E., Rideout, J. R., Dillon, M. R., Bokulich, N. A., Abnet, C. C., Al-Ghalith, G. A., Alexander, H., Alm, E. J., Arumugam, M., Asnicar, F., Bai, Y., Bisanz, J. E., Bittinger, K., Brejnrod, A., Brislawn, C. J., Brown, C. T., Callahan, B. J., Caraballo-Rodríguez, A. M., Chase, J., Cope, E. K., … Caporaso, J. G. (2019). Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nature Biotechnology, 37(8), 852–857. Available online: https://doi.org/10.1038/s41587-019-0209-9.
Caputo, D., Rattansingh, J., Hernandez, V., Nash, B. (2019). An Analysis of Soil Microhabitats in Revolutionary War, Civil War, and Modern Graveyards on Long Island, NY. Journal of Emerging Investigators. Available online: https://emerginginvestigators.org/articles/an-analysis-of-soil-microhabitats-in-revolutionary-war-civil-war-and-modern-graveyards-on-long-island-ny.
CyVerse (2019). DNA Subway: Fast Track to Gene Annotation and Genome Analysis Home Page. Available online at: https://dnasubway.cyverse.org/ (accessed January 5, 2021).
Gray, M. W., Sankoff, D., Cedergren, R. J. (1984). On the evolutionary descent of organisms and organelles: a global phylogeny based on a highly conserved structural core in small subunit ribosomal RNA. Nucleic Acids Res., 12, 5837–5852. Available online: DOI:10.1093/nar/12.14.5837.
He, S., Guo, L., Niu, M., Miao, F., Jiao, S., Hu, T., Long, M. (2017). Ecological diversity and co-occurrence patterns of bacterial community through soil profile in response to long-term switchgrass cultivation. Available online: DOI:10.1038/s41598-017-03778-7.
Knight, R., Vrbanac, A., Taylor, B. C., Aksenov, A., Callewaert, C., Debelius, J., Gonzalez, A., Kosciolek, T., McCall, L. I., McDonald, D., Melnik, A. V., Morton, J. T., Navas, J., Quinn, R. A., Sanders, J. G., Swafford, A. D., Thompson, L. R., Tripathi, A., Xu, Z. Z., Zaneveld, J. R., Dorrestein, P. C. (2018). Best practices for analysing microbiomes. Nature Reviews. Microbiology, 16(7), 410–422. Available online: https://doi.org/10.1038/s41579-018-0029-9.
Maeda, K., Hanajima, D., Toyoda, S., Yoshida, N., Morioka, R., Osada, T. (2011). Microbiology of nitrogen cycle in animal manure compost. Microbial Biotechnology, 4(6), 700–709. Available online: DOI:10.1111/j.1751-7915.2010.00236.x.
MO BIO Laboratories, Inc. (2014). Manual [PDF].
Papale, M., Romano, I., Finore, I., Lo Giudice, A., Piccolo, A., Cangemi, S., Di Meo, V., Nicolaus, B., Poli, A. (2021). Prokaryotic Diversity of the Composting Thermophilic Phase: The Case of Ground Coffee Compost. Microorganisms, 9(2), 218. Available online: DOI:10.3390/microorganisms9020218.
Parada, A. E., Needham, D. M., Fuhrman, J. A. (2016). Every base matters: assessing small subunit rRNA primers for marine microbiomes with mock communities, time series and global field samples. Environ. Micro., 18, 1403–1414. Available online: DOI:10.1111/1462-2920.13023.
Trautmann, Nancy and Olynciw, Elaina. n.d. "CORNELL Composting - Compost Microorganisms." Available online: https://compost.css.cornell.edu/microorg.html.
Yuan, G. N., Marquez, G. P. B., Deng, H., Iu, A., Fabella, M., Salonga, R. B., Ashardiono, F., Cartagena, J. A. (2022). A review on urban agriculture: technology, socio-economy, and policy. Heliyon, 8(11), e11583. Available online: DOI:10.1016/j.heliyon.2022.e11583.